Predicting functional modules: gene families vs taxa
Giulio Benedetti
University of Turkugiulio.benedetti@utu.fi
12 May 2026
Source:vignettes/comparison.Rmd
comparison.RmdPredicting modules from functional profile
# Load gene family experiment
genes <- curatedMetagenomicData(
"SankaranarayananK_2015.gene_families", dryrun = FALSE
)
genes <- genes[[1]]
# Keep only unstratified gene families
genes <- genes[!grepl("|", rownames(genes), fixed = TRUE), ]
# Print experiment
genes
#> class: SummarizedExperiment
#> dim: 1630528 37
#> metadata(0):
#> assays(1): gene_families
#> rownames(1630528): UNMAPPED UniRef90_A5KNK6 ... UniRef90_A0A1B1NI32
#> UniRef90_A0A343UZQ1
#> rowData names(0):
#> colnames(37): CA01 CA02 ... CA40 CA41
#> colData names(23): study_name subject_id ... BMI smoker
graph <- ariadne()
searchPath(graph, uniref90 ~ gmm, k = 3)
#> Path 1:
#> uniref90 -(ChocoPhlAn)-> ko -(GM)-> gmm
#>
#> Path 2:
#> uniref90 -(UniProt)-> kegg_genes -(KEGG)-> ko -(GM)-> gmm
#>
#> Path 3:
#> uniref90 -(UniProt)-> ec -(KEGG)-> ko -(GM)-> gmm
# Weave bindings between taxa and GMMs
uniref2gmm <- weaveComplex(graph, uniref90 ~ gmm, init = rownames(genes))
#> uniref90 -(ChocoPhlAn)-> ko
#> ko -(GM)-> gmm
# Print some results
head(uniref2gmm)
#> uniref90 gmm cov gmm.name
#> 1 UniRef90_A0A068QVT4 MF0090 0.3333333 ethanol production I
#> 2 UniRef90_A0A068QVT4 MF0091 0.5000000 ethanol production II
#> 3 UniRef90_A0A074JIT2 MF0015 1.0000000 fructose degradation
#> 4 UniRef90_A0A075K6L7 MF0067 0.2000000 glycolysis (preparatory phase)
#> 5 UniRef90_A0A075NCB4 MF0005 0.3333333 starch degradation
#> 6 UniRef90_A0A076GS69 MF0097 0.1428571 homoacetogenesis
genes <- addModules(genes, uniref2gmm, as = "names")
uniref.modules <- agglomerateByModule(
genes, by = "rows", group = levels(uniref2gmm$gmm.name)
)
uniref.modules
#> class: SummarizedExperiment
#> dim: 94 37
#> metadata(0):
#> assays(1): gene_families
#> rownames(94): 4-aminobutyrate degradation acetate to acetyl-CoA ...
#> valine degradation I xylose degradation
#> rowData names(0):
#> colnames(37): CA01 CA02 ... CA40 CA41
#> colData names(23): study_name subject_id ... BMI smoker
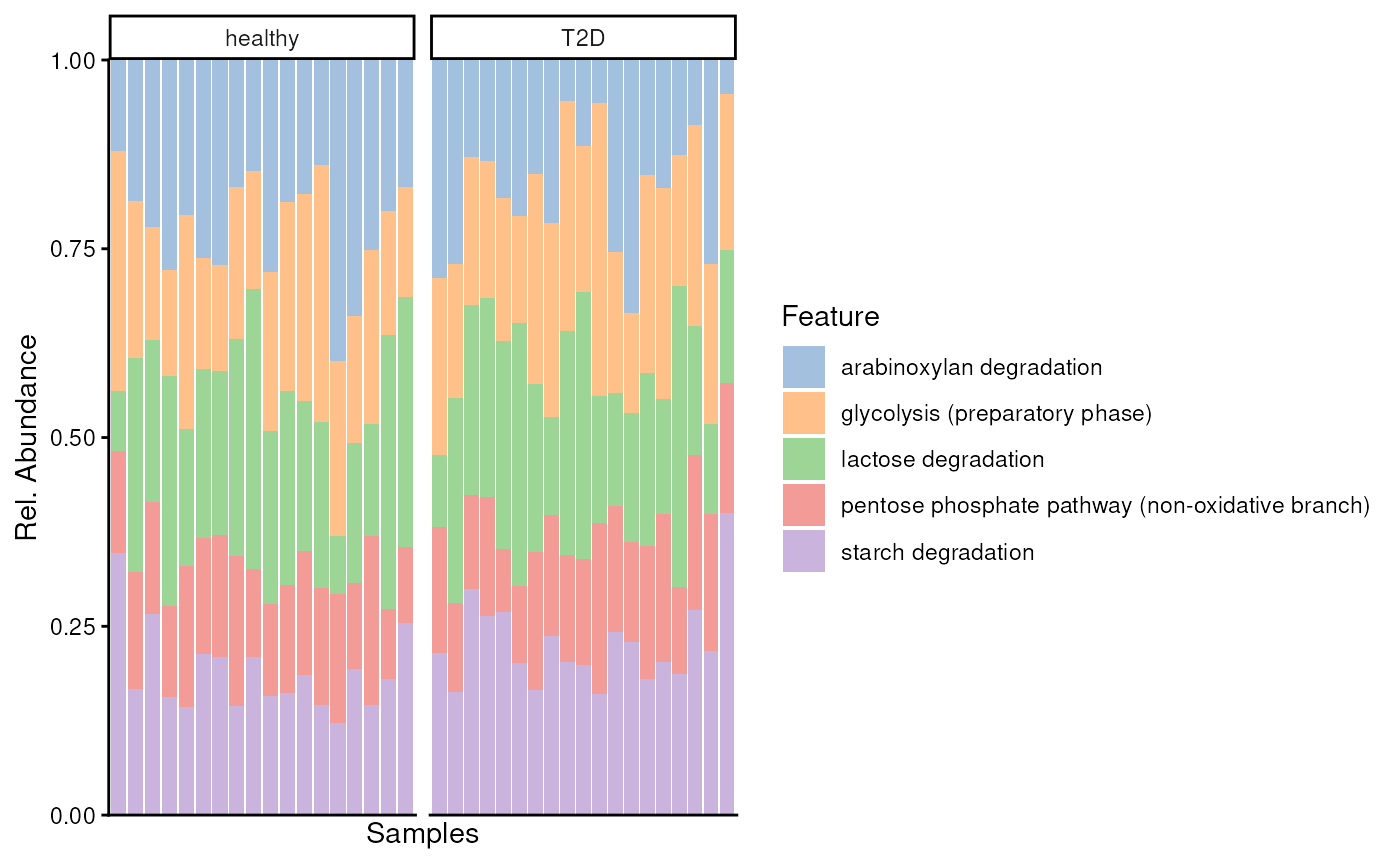
top.mods <- getTop(uniref.modules, assay.type = "gene_families")
plotAbundance(
uniref.modules[top.mods, ],
assay.type = "gene_families",
col.var = "disease",
scales = "free_x",
as.relative = TRUE,
facet.cols = TRUE
)
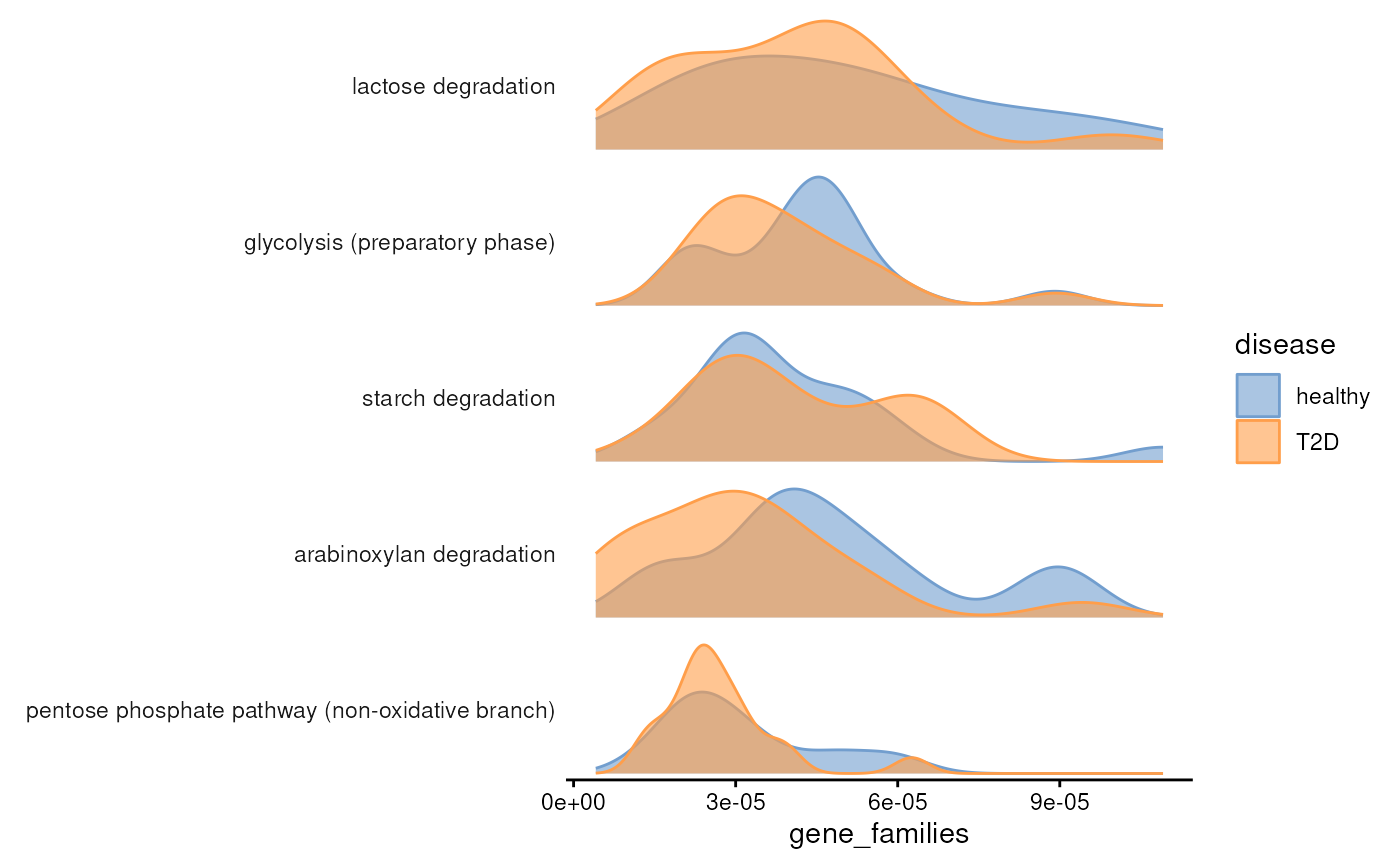
plotAbundanceDensity(
uniref.modules,
assay.type = "gene_families",
n = 5,
layout = "density",
colour.by = "disease"
)
Predicting modules from taxonomic profile
tse <- curatedMetagenomicData(
"SankaranarayananK_2015.relative_abundance",
rownames = "NCBI",
dryrun = FALSE
)
#>
#> $`2021-03-31.SankaranarayananK_2015.relative_abundance`
#> dropping rows without rowTree matches:
#> k__Bacteria|p__Actinobacteria|c__Coriobacteriia|o__Coriobacteriales|f__Coriobacteriaceae|g__Collinsella|s__Collinsella_stercoris
#> k__Bacteria|p__Actinobacteria|c__Coriobacteriia|o__Coriobacteriales|f__Coriobacteriaceae|g__Enorma|s__[Collinsella]_massiliensis
#> k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Carnobacteriaceae|g__Granulicatella|s__Granulicatella_elegans
#> k__Bacteria|p__Proteobacteria|c__Betaproteobacteria|o__Burkholderiales|f__Sutterellaceae|g__Sutterella|s__Sutterella_parvirubra
#> Duplicated labels were made unique.
tse <- tse[[1]]
tse
#> class: TreeSummarizedExperiment
#> dim: 365 37
#> metadata(0):
#> assays(1): relative_abundance
#> rownames(365): species:33039 species:33038 ... species:29388
#> species:76775
#> rowData names(7): superkingdom phylum ... genus species
#> colnames(37): CA01 CA02 ... CA40 CA41
#> colData names(23): study_name subject_id ... BMI smoker
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
#> rowLinks: a LinkDataFrame (365 rows)
#> rowTree: 1 phylo tree(s) (10430 leaves)
#> colLinks: NULL
#> colTree: NULL
searchPath(graph, taxid ~ gmm, k = 3)
#> Path 1:
#> taxid -(UniProt)-> uniref50 -(ChocoPhlAn)-> ko -(GM)-> gmm
#>
#> Path 2:
#> taxid -(UniProt)-> uniref90 -(ChocoPhlAn)-> ko -(GM)-> gmm
#>
#> Path 3:
#> taxid -(UniProt)-> uniref50 -(ChocoPhlAn)-> uniref90 -(ChocoPhlAn)-> ko -(GM)-> gmm
# Link taxa to gut metabolic modules (remove taxid 562 to speed up)
tax2gmm <- weaveComplex(graph, taxid ~ gmm, k = 2, init = rowData(tse)$species)
#> taxid -(UniProt)-> uniref90
#> uniref90 -(ChocoPhlAn)-> ko
#> ko -(GM)-> gmm
head(tax2gmm)
#> taxid gmm cov gmm.name
#> 1 105841 MF0036 0.1666667 isoleucine degradation
#> 2 105841 MF0041 0.1428571 valine degradation I
#> 3 105841 MF0057 0.1666667 lysine degradation I
#> 4 105841 MF0067 0.2000000 glycolysis (preparatory phase)
#> 5 105841 MF0088 0.3333333 butyrate production I
#> 6 105841 MF0089 0.5000000 butyrate production II
tse <- addModules(tse, tax2gmm, key = "species", as = "names")
tax.modules <- agglomerateByModule(
tse, by = "rows", group = levels(tax2gmm$gmm.name)
)
tax.modules
#> class: TreeSummarizedExperiment
#> dim: 99 37
#> metadata(0):
#> assays(1): relative_abundance
#> rownames(99): 4-aminobutyrate degradation acetate to acetyl-CoA ...
#> valine degradation I xylose degradation
#> rowData names(0):
#> colnames(37): CA01 CA02 ... CA40 CA41
#> colData names(23): study_name subject_id ... BMI smoker
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
#> rowLinks: NULL
#> rowTree: NULL
#> colLinks: NULL
#> colTree: NULL
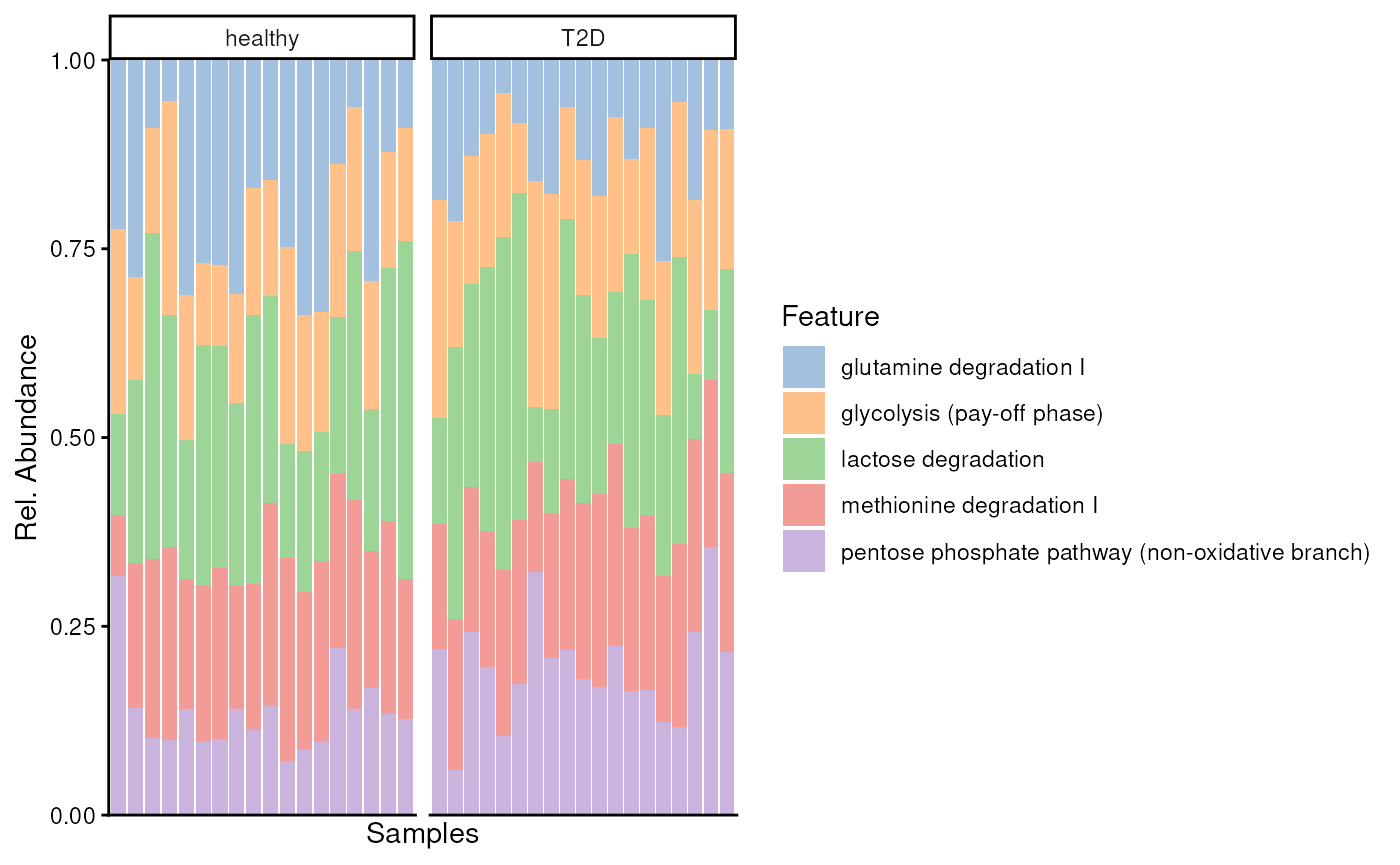
top.mods <- getTop(tax.modules, assay.type = "relative_abundance")
plotAbundance(
tax.modules[top.mods, ],
assay.type = "relative_abundance",
col.var = "disease",
scales = "free_x",
as.relative = TRUE,
facet.cols = TRUE
)
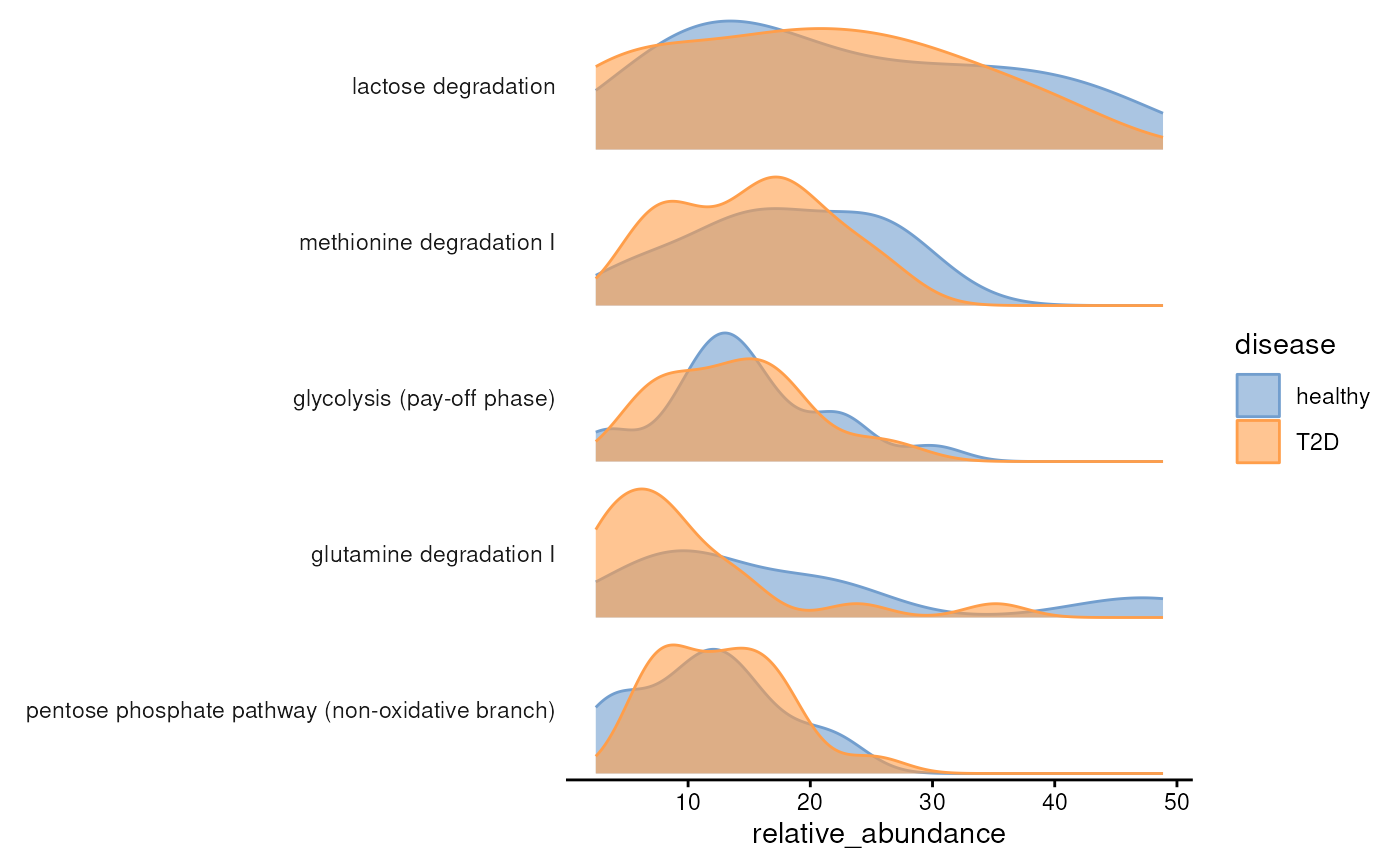
plotAbundanceDensity(
tax.modules,
assay.type = "relative_abundance",
n = 5,
layout = "density",
colour.by = "disease"
)
Correlating results
mae <- MultiAssayExperiment(
experiments = ExperimentList(uniref = uniref.modules, tax = tax.modules)
)
cor <- getCrossAssociation(
mae,
assay.type1 = "gene_families",
assay.type2 = "relative_abundance",
sort = TRUE
)
#> Calculating correlations...
#> altexp1: -, altexp2: -, assay.type1: gene_families, col.var1: -, dimred1: -, assay.type2: relative_abundance, col.var2: -, dimred2: -
#> by: 1, function: stats::cor, method: kendall, test.signif: FALSE, p.adj.method: -, paired: FALSE, show.warnings: TRUE
#> Sorting results...
cor$Var1 <- as.character(cor$Var1)
cor$Var2 <- as.character(cor$Var2)
cor <- cor[cor$Var1 == cor$Var2, ]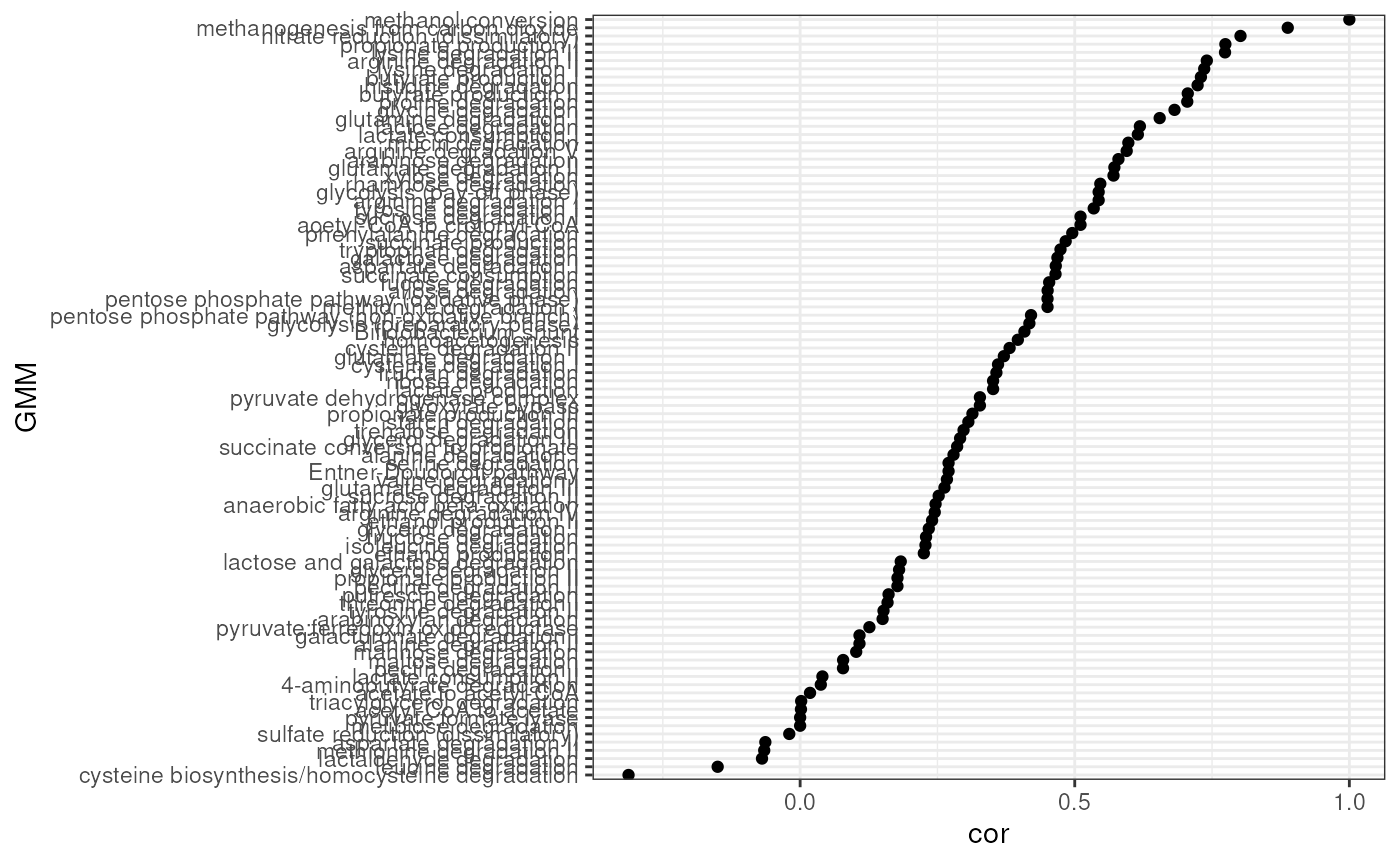
Reproducibility
R session information:
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] miaViz_1.21.0 ggraph_2.2.2 ggplot2_4.0.3
#> [4] mia_1.19.8 MultiAssayExperiment_1.39.0 curatedMetagenomicData_3.19.0
#> [7] TreeSummarizedExperiment_2.21.0 Biostrings_2.81.1 XVector_0.53.0
#> [10] SingleCellExperiment_1.35.0 SummarizedExperiment_1.43.0 Biobase_2.73.1
#> [13] GenomicRanges_1.65.0 Seqinfo_1.3.0 IRanges_2.47.0
#> [16] S4Vectors_0.51.1 BiocGenerics_0.59.0 generics_0.1.4
#> [19] MatrixGenerics_1.25.0 matrixStats_1.5.0 ariadne_0.2.3
#> [22] BiocStyle_2.41.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.6.0 MultiFactor_0.1.2 ggplotify_0.1.3 filelock_1.0.3
#> [5] tibble_3.3.1 polyclip_1.10-7 XML_3.99-0.23 DirichletMultinomial_1.55.0
#> [9] lifecycle_1.0.5 httr2_1.2.2 vroom_1.7.1 lattice_0.22-9
#> [13] MASS_7.3-65 SnowballC_0.7.1 magrittr_2.0.5 sass_0.4.10
#> [17] rmarkdown_2.31 jquerylib_0.1.4 yaml_2.3.12 otel_0.2.0
#> [21] DBI_1.3.0 RColorBrewer_1.1-3 abind_1.4-8 purrr_1.2.2
#> [25] yulab.utils_0.2.4 tweenr_2.0.3 rappdirs_0.3.4 gdtools_0.5.0
#> [29] ggrepel_0.9.8 tokenizers_0.3.0 irlba_2.3.7 tidytree_0.4.7
#> [33] rentrez_1.2.4 vegan_2.7-3 pkgdown_2.2.0 permute_0.9-10
#> [37] DelayedMatrixStats_1.35.0 codetools_0.2-20 DelayedArray_0.39.1 scuttle_1.23.0
#> [41] ggforce_0.5.0 tidyselect_1.2.1 aplot_0.2.9 farver_2.1.2
#> [45] ScaledMatrix_1.21.0 viridis_0.6.5 BiocFileCache_3.3.0 jsonlite_2.0.0
#> [49] BiocNeighbors_2.7.2 decontam_1.33.0 tidygraph_1.3.1 scater_1.41.1
#> [53] systemfonts_1.3.2 tools_4.6.0 ggnewscale_0.5.2 progress_1.2.3
#> [57] treeio_1.37.0 ragg_1.5.2 Rcpp_1.1.1-1.1 glue_1.8.1
#> [61] gridExtra_2.3 SparseArray_1.13.2 xfun_0.57 mgcv_1.9-4
#> [65] dplyr_1.2.1 withr_3.0.2 BiocManager_1.30.27 fastmap_1.2.0
#> [69] bluster_1.23.0 digest_0.6.39 rsvd_1.0.5 R6_2.6.1
#> [73] gridGraphics_0.5-1 textshaping_1.0.5 RSQLite_3.52.0 ecodive_2.2.6
#> [77] tidyr_1.3.2 fontLiberation_0.1.0 data.table_1.18.4 DECIPHER_3.9.0
#> [81] prettyunits_1.2.0 graphlayouts_1.2.3 httr_1.4.8 htmlwidgets_1.6.4
#> [85] S4Arrays_1.13.0 pkgconfig_2.0.3 gtable_0.3.6 blob_1.3.0
#> [89] S7_0.2.2 janeaustenr_1.0.0 htmltools_0.5.9 fontBitstreamVera_0.1.1
#> [93] bookdown_0.46 scales_1.4.0 png_0.1-9 ggfun_0.2.0
#> [97] knitr_1.51 tzdb_0.5.0 reshape2_1.4.5 rncl_0.8.9
#> [101] nlme_3.1-169 curl_7.1.0 cachem_1.1.0 stringr_1.6.0
#> [105] BiocVersion_3.24.0 parallel_4.6.0 vipor_0.4.7 arrow_24.0.0
#> [109] AnnotationDbi_1.75.0 desc_1.4.3 pillar_1.11.1 grid_4.6.0
#> [113] vctrs_0.7.3 BiocSingular_1.29.0 dbplyr_2.5.2 beachmat_2.29.0
#> [117] cluster_2.1.8.2 beeswarm_0.4.0 evaluate_1.0.5 readr_2.2.0
#> [121] cli_3.6.6 compiler_4.6.0 rlang_1.2.0 crayon_1.5.3
#> [125] tidytext_0.4.3 labeling_0.4.3 forcats_1.0.1 plyr_1.8.9
#> [129] fs_2.1.0 ggbeeswarm_0.7.3 ggiraph_0.9.6 stringi_1.8.7
#> [133] viridisLite_0.4.3 BiocParallel_1.47.0 assertthat_0.2.1 lazyeval_0.2.3
#> [137] fontquiver_0.2.1 Matrix_1.7-5 ExperimentHub_3.3.0 patchwork_1.3.2
#> [141] hms_1.1.4 sparseMatrixStats_1.25.0 bit64_4.8.0 KEGGREST_1.53.0
#> [145] AnnotationHub_4.1.0 igraph_2.3.1 memoise_2.0.1 bslib_0.10.0
#> [149] rotl_3.1.1 ggtree_4.3.0 bit_4.6.0 ape_5.8-1